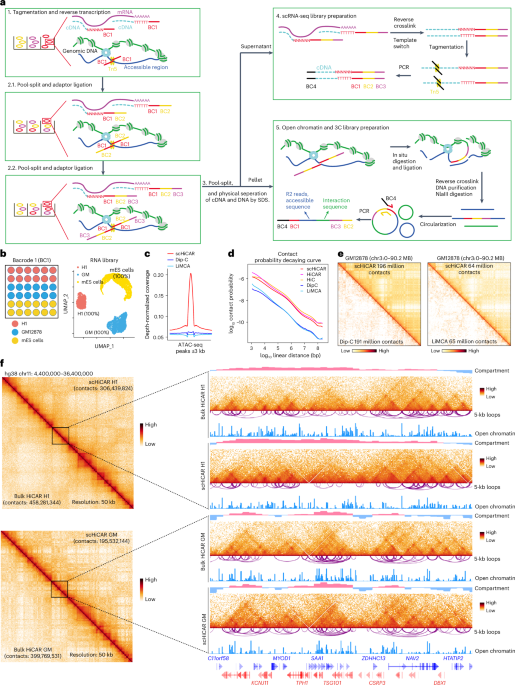
a. Proportion of CRE–gene pairs that cross topologically associating domain (TAD) boundaries. Comparisons were made between pairs predicted by BICCN snATAC-seq based on co-accessibility (light blue) and pairs identified by scHiCAR chromatin interactions (dark blue) across different brain cell types. For fair comparison, the number of co-accessible pairs (n = 418,528) was comparable to the number of scHiCAR pairs (n = 440,710) by selecting top-ranked pairs by co-accessibility score. b. Enrichment of VISTA-validated enhancers among different sets of distal cCREs. Compared were: (i) snATAC-seq distal cCREs linked to gene promoters by the ABC model (ABC score > 0.05) and (ii) snATAC-seq distal cCREs linked by scHiCAR chromatin interactions. Odds ratios were calculated using Fisher’s exact test. For fair comparison, both sets contained similar numbers of cCREs (47,332 for ABC and 43,290 for scHiCAR). c. Genome browser view showing a VISTA-validated enhancer (mm1451.0) that interacts with the Grk3 promoter in astrocytes. Chromatin interactions called by scDeepLUCIA are shown as black squares on the contact matrix. d. Log-normalized expression of genes with promoter accessibility linked to cCREs (red) versus unlinked genes (pink) across brain cell types. Box plots indicate the median, 25th–75th percentiles, and 1.5× IQR whiskers. P-values from two-sided unpaired Wilcoxon tests. The number of samples from left to right are 7,385; 5,066; 7,275; 4,990; 7,269; 5,227; 5,477; 4,212; 7,327; 4,501; 7,616; 5,143; 6,731; 5,012; 5,555; 3,756; 7,269; 4,710; 7,020; 5,135; 6,551; 5,435; 2,009; 2,448; 2,243; 1,708; 7,361; 4,609; 7,857; 4,859; 6,211; 4,109; 1,901; 1,611; 6,548; 5,473; 7,136; 5,421; 5,748; 5,960; 5,741; 5,707; 4,861; 5,419. The p-values are 1.37e-38, 2.45e-35, 6.65e-41, 3.05e-22, 1.03e-30, 7.47e-40, 1.32e-37, 2.78e-14, 1.89e-27, 1.88e-47, 2.10e-50, 1.01e-19, 0.00024, 3.78e-24, 6.45e-24, 6.14e-19, 3.23e-10, 2.21e-36, 4.91e-28, 2.85e-33, 7.86e-54, 1.04e-28. e. An example of positive association between gene expression and the number of scHiCAR chromatin interactions in L23IT-1 neurons. Shown are RNA expression (red) and chromatin accessibility (blue) tracks. f. Scatterplot showing correlation between changes in the number of scHiCAR chromatin interactions and changes in gene expression between two brain cell types. The dashed line indicates the linear regression fit. The Pearson correlation coefficient (PCC) and associated p-value were calculated using a two-sided Pearson correlation test. g. Density plot showing the distribution of Pearson correlation coefficients (PCCs) between gene expression and chromatin accessibility at distal CREs for all CRE–gene pairs. A subset of 20,270 pairs with the strongest positive correlations (FDR < 0.05) was defined as high-confidence enhancer–gene pairs (red). Randomized pairings (gray) serve as a control. h. Comparison of enhancer–gene correlation scores between scHiCAR and Paired-seq. Top 34,473 enhancer–gene pairs predicted by scHiCAR were compared with the same number of pairs from Paired-seq based on mRNA and H3K27ac signal correlation. Distribution of Spearman correlation coefficients is shown. P-value calculated by two-sided unpaired Wilcoxon test.
Source data
— Source: Nature Biotechnology (https://www.nature.com/articles/s41587-026-03013-7)